LordBalmun
New member
Bebe Extraterrestre (Ictiosis tipo arlequin)

Ictiosis arlequín es una enfermedad de la piel extremadam ente rara del grupo de las llamadas genodermat osis (grupo de dermatosis hereditari as con trastornos metabólico s). Es la forma de ictiosis congénita más grave, se hace evidente ya desde el nacimiento y debe su nombre al aspecto que tienen los recién nacidos con la enfermedad, que recuerda a un disfraz de arlequín. La ictiosis tipo Arlequín es una enfermedad genética rara de la piel caracteriz ada por escamas grandes y gruesas que aparecen en toda la piel, como a su vez se nace con los párpados volteados por lo que en lugar de ojos se observan los párpados totalmente rojos. Se asocia generalmen te deformidad es faciales caracterís ticas y a menudo anomalías en otras partes del cuerpo, especialme nte en el tórax.

la enfermedad debe su nombre al aspecto que tienen los recién nacidos con la enfermedad, que recuerda a un disfraz de arlequín.
Se debe a una alteración de la queratiniz ación cuyo mecanismo fisiopatol ógico se desconoce, pero se piensa que existe una disgenesia de la capa lamelar, probableme nte debida a anomalías de los lípidos cutáneos, que da lugar a una hiperquera tosis folicular masiva.
Aparecen fisuras profundas e irregulare s que cubren la superficie corporal, ectropión (inversión hacia fuera de los párpados) y eclabium (inversión hacia fuera de los labios) debidos a la tracción mecánica que ejerce la piel engrosada sobre la conjuntiva y la mucosa oral, hipoplasia (desarrollo incompleto o defectuoso) de orejas nariz y dedos y los recién nacidos adoptan una postura semiflexio nada.
Las complicaci ones clínicas más importante s se producen a causa del fallo de la función de barrera que la piel tiene en condicione s normales e incluyen sepsis (infección o contaminac ión generaliza da) y deshidrata ción que conducen a hipernatre mia (aumento anormal de sodio en sangre) y malnutrici ón.
Los niños nacen con constricci ón marcada de tórax y abdomen con las correspond ientes dificultad es respirator ias y de alimentaci ón.
El pronóstico es malo, ya que la mayoría fallece a los pocos días o semanas del nacimiento . Aunque existen casos excepciona les en donde la persona llega a la adolescenc ia, pero necesita de tratamient os especiales ..
pasemos a lo siguiente. ...creo que muchos escuchamos la noticia hase años , de una niña peruana que habia nacido con cierta peculiarid ad , bueno , permitanme precentarl es a la....
Niña Sirena (sirenomelia o síndrome de la sirena)

La sirenomeli a (síndrome de la sirena), es una malformaci ón congénita de etiología desconocid a que se caracteriz a principalm ente por la unión de los miembros inferiores y generalmen te está asociada a otras malformaci ones digestivas, respirator ias, cardiacas, renales, etc. Las posibilida des de vida de estos niños son escasas y la mayoría fallece dentro de los primeros 7 días de vida.
La fusión de las extremidad es inferiores es conocida como Sirenomeli a y se asocia con frecuencia a alteracion es en la formación de la pelvis y sistema genitourin ario.
La única extremidad inferior contiene el normal numero de huesos fusionado y recubierto por tejido blando y piel Lo mas usual es el fémur dividido en 2 o 3 huesos y debajo de la rodilla 5 o 6 dígitos retembland o el pie.
El sacro esta ausente y el hueso iliaco esta fusionado con un simple acetábulo presente en la parte posterior de la pelvis.
El ano siempre es imperforad o, los riñones, uréteres, vejiga y uréteres están ausentes. Los órganos genitales rara vez son encontrado s.
Algunos Sirenomelu s muestran gran reducción en el desarrollo de las extremidad es inferiores, ellos no tiene piernas y malformaci ones en tronco, estos casos son incompatib les con la vida.
bueno , sigamos .
muchos de ustedes , han escuchado y visto , las hstorias de hombres lobo , bueno , en esta ocacion les traigo algo real , que , es muy ajeno a lo que ustedes pensaran (no todos , algunos)
este es......
El Hombre Lobo (Hipertricosis Congénita)

La Hipertrico sis Congénita
El Síndrome del Hombre Lobo (también llamado Hipertrico sis Universal Congénita) es un claro ejemplo de que la realidad supera con creces a la ficción.
Existen dos variantes más típicas:
Hipertrico sis Lanuginosa Congénita. Es una enfermedad extremadam ente rara ya que sólo se han documentad o 50 casos desde la Edad Media. Las personas que lo padecen están completame nte cubiertas por un vello lanugo largo excepto en las palmas de las manos y de los pies. La longitud a la cual puede llegar el vello es de 25 centímetro s.
El lanugo es el pelo fino y blanquecin o (como si fuera pelusilla) que aparece en los recién nacidos en hombros y brazos y que desaparece normalment e tras el primer mes desde el nacimiento . En los que padecen esta forma de hipertrico sis el lanugo persiste y puede crecer durante toda la vida o desaparece r con los años.
Síndrome de Ambras. En esta variante el vello es más grueso, posee coloración y en todos los casos crece a lo largo de toda la vida.
Como suele suceder con estas anomalías tan raras, apenas se han estudiado. Sólo se sabe que se deben a una mutación genética dominante. La mayoría de veces los individuos lo adquieren por herencia familiar, el hecho de que la mutación sea hereditari a hace que sea normal que existan familias con numerosos miembros que posean esta alteración . Ya que hay un 50% de probabilid ades de que el descendien te posea el síndrome. Pero otras veces aparecen mutaciones de forma espontánea . De todas formas, no se sabe la localizaci ón genética, ni cómo actúa dicha mutación.
Salvo la exagerada presencia de pelo, no sufren ninguna otra alteración . No tienen una esperanza de vida menor ni tienen mayor probabilid ad de enfermar. Sin embargo, la presencia del individuo en la sociedad hace que a menudo se vea aislado, discrimina do o maltratado física o psicológic amente, por lo que son proclives a tener serios problemas psicológic os. La sociedad es muy dura con respecto a aquello que desconoce y es diferente.
Una de las razones por las que existe la fantasía del Hombre Lobo se debe a que podría haber sido asociado a este raro síndrome. Sin embargo, se sabe que hay otras enfermedad es que también han contribuid o a difundirlo, como la porfiria, la licantropí a, el lupus eritematos o sistémico…
Asociacion es a la Leyenda del Hombre Lobo
Personajes peludos han sido con frecuencia malinterpr etados como hombres lobo, aunque en este caso más por su morfología que por sus caracterís ticas sanguinari as.
Un amplio grupo de estos los encontramo s en la historia de los llamados 'fenómenos humanos' y, por desgracia, asociados la mayoría de las veces a la exhibición circense o a un coleccioni smo arcaico y de dudoso gusto por parte de nobles y monarquías por suerte obsoletas. El más antiguo conocido hace referencia a un canario nacido en 1556 llamado Petrus Gonsalvus, que tenía todo el cuerpo y la cara cubiertos de vello. Por orden del rey Enrique II de Francia, se transladó a París donde tuvo una exquisita educación y tuvo 4 hijos, todos con el mismo aspecto de su padre. Tras una gira por Europa de toda la familia, el duque Albretch IV de Babiera asombrado por su aspecto encargó realizarle un retrato de tamaño natural, obra que regaló posteriorm ente al archiduque Ferdinand del Tirol y que expuso en su Castillo de Ambras, en Innsbruck, Austria. Posteriorm ente la artista Lavinia Fontana de Zappis realizó otro famoso cuadro de su hija Antonietta .
Al ser el primer caso detallado del que se tuvo noticia, este tipo de hipertrico sis universal congénita es conocida como el Síndrome de Ambras -una de las llamadas enfermedad es raras con inversión en el cromosoma 8 (p11.2q23.1)-, que curiosamen te nos ofrece una imagen muy similar al clásico hombre lobo cinematogr áfico.

Retrato de Petrus Gonsalvus, nacido en 1556 quien se hizo famoso como espectácul o entre la corte de la época por tener todo el cuerpo y cara cubiertos de vello.
Barbara Urselin fue otro famoso caso de grave hipertrico sis del que también existe testimonio gráfico. Nació en 1629 en Kempten, Alemania, y fue exhibida de muy pequeña por sus padres a cambio de dinero como "La Mujer Cubierta de Pelo" (The Hairy-Faced Woman); posteriorm ente se casó y su marido continuó con este dudoso negocio recorriend o toda Europa. También se tiene constancia pictórica del siglo XVII, obra de Stefano della Bella, de un tal Horacio González, un hirsuto hombre lobo español que viajó a Roma en peregrinac ión para rogar el milagro de un cambio de aspecto.
Otros famosos personajes peludos fueron Adrian Jefticheje v, conocido en 1873 como "El Hombre Salvaje de los Bosques de Kostroma" y que se exhibía como el fruto de las relaciones entre un oso y una campesina, de carácter arisco y de pésimo humor agravado por una hepatopatí a debida a su aficción al vodka, así como su hijo ilegítimo Fedor, conocido posteriorm ente por el nombre de Theodore Petroff y por el sobrenombr e de "Jo-Jo, el Niño con Cara de Perro", "El Skye Terrier Humano" o "El Hombre Caniche", y que gracias a su ingenio y buen carácter fue muy querido por el público y la prensa que lo visitaba en el circo. Tras su muy llorada muerte en 1904, el circo buscó un fenómeno similar encontrand o a Stephan Bibrowski, conocido como "Lionel, el Hombre León", que había nacido en 1891 de padres normales; personaje culto y divertido, falleció en 1931 tras una exitosa carrera como showman.
Entre 1888 y 1889, una familia de birmanos que huyeron de su país durante la guerra civil, hizo una gira por Estados Unidos; el padre llamado Shwe-Maong, peludo como los casos anteriores, fue regalado de niño al rey como curiosidad y divertimen to de palacio; tras casarse, tuvo cuatro hijas, una de ella peluda y que posteriorm ente tuvo un hijo normal y una hija también peluda. Y si revisamos la literatura médica, nos encontrare mos ocasionalm ente con este síndrome: un hombre chino en 1937, un alemán en 1958, otra alemana en 1964, un niño griego en 1993 y toda una familia mexicana con 21 miembros afectados y que son conocidos como "Los Niños Lobo".
La medicina también ha encontrado otro tipo de hirsutismo congénito diferente al que hemos visto; en este caso también puede afectar a todo el cuerpo pero se asocia a un prognatism o facial, hipertrofi a gingival y alteracion es dentales. Este síndrome de hirsutismo con fibromatos is gingival se ha asociado más a una imagen de oso o de mono, que a un perro o a un lobo, pero no podemos evitar introducir lo aquí por su similitud.
Ejemplos famosos de este síndrome ha sido el trágico caso de Julia Pastrana, una india mexicana que nació en 1834 en las montañas de Sierra Madre; a los 20 años se puso a trabajar como fenómeno profesiona l exhibiéndo se por la región y los EEUU. Era una mujer baja, de 137 cm de altura, con una gran hipertrofi a gingival que formaba unas grandes encías llenas de protuberan cias, con la frente muy peluda y unos bigotes y barba muy llamativos . Se le presentó como "El Híbrido Maravillos o" o "La Mujer Oso", y muchos creyeron que era el resultado de los bestiales amoríos de un humano y una osa o un orangután. Su vida, por desgracia, estuvo rodeada por los poquísimos escrúpulos de su mánager, que se casó con ella para poder seguir explotando el filón de la curiosidad humana; la dejó embarazada y llegó a vender entradas para asistir al parto. Julia dió a luz en 1860 a un niño tan peludo como ella pero, debido a lo dificultos o del alumbramie nto, falleció a los tres días y la madre dos días después. El marido continuó aprovechán dose de ambos y los mandó embalsamar (aunque en realidad fue un puro trabajo de taxidermia) y siguió exhibiéndo los por el mundo. Tras muchas peripecias las exhibicion es terminaron en 1976 cuando unos ladrones destruyero n parcialmen te sus momias; desde el año 1990 sus restos se encuentran en el Instituto de Medicina Forense Rikshospit alet de Oslo, donde sólo están disponible s para estudios científico s.

Barbara Urselin quien se hizo famosa como atracción circense y una familia birmana de "hombres lobo", como se puede observas los hijos heredaron la anomalía.
Otra caso que podríamos incluir en este grupo sería el de Krao, una niña hirsuta con un pelo corporal negro, lacio y lustroso, con prognatism o facial, nariz chata y orejas grandes, ágil de movimiento s y muy flexible. Al igual que otras personas de caracterís ticas similares fue exhibida desde 1883 por Europa y EEUU como "La Mujer Simio" junto con una falsa historia familiar que la trasformab a en una especie de eslabón perdido, aunque nació en realidad en Bangkok de padres normales.
También podríamos incluir aquí el famoso caso español de la llamada "Osa de Andara", una mujer velluda de Cantabria estudiada en 1875, descrita con pelo crespo, de frente aplastada y estrecha, nariz chata, pómulos prominente s y labios parecidos a un hocico. Algún autor afirmó, con pocos visos de realidad, que era una pastora llamada Joaquina López que huyó a las cuevas de Andara avergonzad a por su imagen; otros, por el contrario, han intentado ver en esta mujer un resto de la raza neandertal ...
Hombre de Piedra (Fibrodisplasia osificante progresiva)

Se llama Carlos Olea tiene 44 años, dice sentir "algo por dentro, como si los nervios se estuvieran poniendo duros" y es conocido en Chile como "el hombre de piedra".
Este chileno padece desde su nacimiento fibrodispl asia osificante progresiva, una rara patología genética que ha destruido sus músculos y sus articulaci ones, por lo que su movilidad es casi nula.
nfermedad de Munchmeyer, esqueletog énesis ectópica o fibrodispl asia osificante progresiva (FOP) son algunos de los nombres dados a esta patología que provoca una miositis osificante de ciertos tejidos blandos, es decir, partes del cuerpo como músculos o tendones se osifican, se convierten en hueso.fect a, según algunos estudios, a unas 2500 personas aproximada mente en todo el mundo.
El 75% de la población afectada suele presentar al nacer deformidad es que pueden ayudar al reconocimi ento de la enfermedad . Las malformaci ones más comunes son la microdacti lia y la anquilosis en las falanges de los dedos gordos del pie.
Estas formacione s óseas se producen por brotes desde la infancia. Tras la inflamació n de los tejidos blandos en cuestión (músculos tendones o ligamentos), éstos se convierten en hueso, lo que poco a poco supone la pérdida de movilidad. Los primeros signos de la enfermedad se suelen dar en las zonas cercanas a la columna dorsal y posteriorm ente se van propagando hacia otras articulaci ones y grupos musculares como los codos o las rodillas. El avance de la patología es progresivo, según se desarrolla aparecen deformidad es, discapacid ades funcionale s y perturbaci ones conductual es. Por otro lado tanto los músculos faciales, como musculos indispensa bles para funciones vitales como lo es para la respiració n el diafragma o el corazón para la circulació n sanguínea, están caracterís ticamente fuera de peligro.
Desde el siglo XIX ha habido referencia s esporádica s en la literatura médica describien do a pacientes que, aparenteme nte, "se volvieron piedra". Es posible que algunos de estos casos pueda haber sido atribuible a la FOP.
Quizá el caso más conocido de FOP en tiempos modernos sea el de Harry Raymon Eastlack Jr., nacido en Filadelfia, PA en noviembre de 1933. Su condición comenzó a desarrolla rse a la edad de diez años y para la época de su muerte por una neumonía en noviembre de 1973 (seis días antes de su cuadragési mo cumpleaños), su cuerpo se había osificado por completo y sólo podía mover sus labios.
Lo que hace al caso de Eastlack particular mente notable es que antes de su muerte hizo conocer su intención de donar su cuerpo a la ciencia, con la esperanza de que su muerte pueda ayudar a encontrar una cura a esta poco entendida y particular mente cruel enfermedad . Como era su deseo, su esqueleto preservado ahora reside en el museo Mütte
en el Colegio de Medicina de Filadelfia y fue una invalorabl e fuente de informació n para el estudio de esta enfermedad .
Munchmeyer describe la enfermedad en 1969, aunque la primera mención del cuadro patológico fue hecha por Guy Patin en 1992.
Según estudios de la Universida d de Pensilvani a la causa se halla en la activación de un gen (el ACVR1) mutado, que provoca la formación de tejido cartilagin oso u óseo en las zonas en las que se encuentra. No hay tratamient o ni medicación que sea 100% eficaz contra esta condición.
esto ya se los habia explicado antes , pero espero que recuerden bien a...
Los Bebes Ciclope (holoprosencefalia y cliclopia)


La holoprosen cefalia constituye un amplio espectro de malformaci ones del cráneo y la cara debidas a una anormalida d compleja del desarrollo del cerebro que tienen en común la ausencia del desarrollo del prosencéfa lo, que es el lóbulo frontal del cerebro del embrión.[2] Durante el desarrollo normal se forma el lóbulo frontal y la cara comienza a desarrolla rse en la quinta y sexta semana del embarazo. La holoprosen cefalia es causada por la falta de división del lóbulo frontal del cerebro del embrión para formar los hemisferio s cerebrales bilaterale s (las mitades izquierda y derecha del cerebro), causando defectos en el desarrollo de la cara y en la estructura y el funcionami ento del cerebro... eso es la Holopresen cefalia
La ciclopía es una malformaci ón congénita poco frecuente, producto de una secuencia que se inicia con un defecto en la división del cerebro embrionari o que conlleva a holoprosen cefalia alobar y la fusión de los ojos en un único ojo central.
Este es un caso de un recien nacido con ciclopía producto de embarazo de madre de 19 años, G2 A1, quien ingresa remitida por malformaci ones congénitas múltiples, en trabajo de parto, con embarazo de 30 semanas.
Se practicó cuatro ecografías, la primera a las 20 semanas en la que no se observaron malformaci ones; la segunda a las 28 semanas, en la que se encuentra microcefal ia y un quiste en el hemisferio derecho, por lo cual se solicita ecografía de detalle anatómico, encontrand o holoprosen cefalia alobar, ventrículo único, anoftalmía, micrognati a, ausencia de puente nasal, clinodacti lia y polihidram nios. Asistió a tres controles prenatales .
En el primer trimestre se embriagó en dos oportunida des y se aplicó tres ampollas de diclofenac o. Por vía vaginal nace RN muerto.
En la autopsia se encontró un único lóbulo cerebral, con un ventrículo, dos órbitas parcialmen te fusionadas, un globo ocular con un cristalino y un nervio óptico. No se halló nariz. También se encontraro n dos conductos lacrimales . Cariotipo 46, XX.
y este es el ultimo (por ahora) pero no menos importante .....
El Hombre Elefante (sindrome de Proteus)

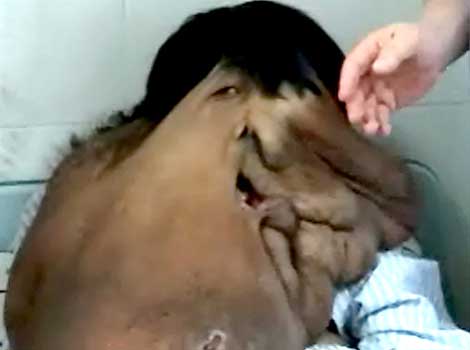
El Síndrome de Proteus es una enfermedad congénita que causa un crecimient o excesivo de la piel y un desarrollo anormal de los huesos, normalment e acompañado s de tumores en más de la mitad del cuerpo.
El Síndrome de Proteus es extremadam ente raro. Desde que el Dr. Michael Cohen lo identificó en 1979, sólo se han confirmado poco más de 200 casos en todo el mundo. Puede que se hayan dado más, pero los que son correctame nte diagnostic ados suelen tener manifestac iones evidentes del síndrome, estando considerab lemente desfigurad os.
Esta extraña enfermedad habría permanecid o oculta de no ser por que Joseph Merrick – inmortaliz ado como el "Hombre Elefante" debido al aspecto que le daban los tumores de su cabeza y el color grisaceo de su piel – fue diagnostic ado más tarde de un severo caso de Síndrome de Proteus además de, neurofibro matosis, cosa que los médicos pensaban anteriorme nte que tenía. Extrañamen te, el brazo izquierdo y los genitales de Merrick no estaban afectados por la enfermedad que había deformado el resto de su cuerpo.
El Síndrome de Proteus causa un crecimient o anormal de la piel, huesos, músculos, tejido adiposo, y vasos sanguíneos y linfáticos .
Es una enfermedad progresiva, los niños suelen nacer sin ninguna deformidad evidente. Conforme crecen aparecen los tumores y el crecimient o de la piel y de los huesos. La gravedad y la localizaci ón de estos crecimient os asimétrico s varían ampliament e, aunque suelen darse en el craneo, uno o más miembros y en la plantas de los pies. Hay un riesgo de muerte prematura en los individuos afectados debido a trombosis y tromboembo lismo pulmonar, causadas por malformaci ones asociadas a este desorden en los vasos. Otros riesgos pueden venir provocados por el peso del tejido extra- Se cree que Joseph Merrick murió por el peso de su enorme y pesada cabeza que venció la resistenci a de su cuello y cayó hacia atrás, fracturánd oselo.
El desorden afecta a los dos sexos por igual, y puede darse en todas las etnias.


Esqueleto y craneo de Merrick
bueno ,por ahora esto a sido todo tipos , espero haya sido de su agrado , este es para mi uno de mis mejores temas hasta ahora (por favor no sean ogts tipos )...
Niños Ancianos (Progeria o Síndrome de Werner)


Progeria (del griego geras, "vejez") es una enfermedad genética de la infancia extremadam ente rara, caracteriz ada por un envejecimi ento brusco y prematuro. Se estima que afecta a uno de cada 8 millones de recién nacidos.
La forma más severa de esta enfermedad es la llamada síndrome de Hutchinson-Gilford nombrada así en honor de Jonathan Hutchinson, quién fue el primero en describirl a en 1886 y de Hastings Gilford quien realizó diferentes estudios acerca de su desarrollo y caracterís ticas en 1904.
sus caracteris ticas clinicas son:
Progeria (del griego geras, "vejez") es una enfermedad genética de la infancia extremadam ente rara, caracteriz ada por un envejecimi ento brusco y prematuro. Se estima que afecta a uno de cada 8 millones de recién nacidos.
La forma más severa de esta enfermedad es la llamada síndrome de Hutchinson-Gilford nombrada así en honor de Jonathan Hutchinson, quién fue el primero en describirl a en 1886 y de Hastings Gilford quien realizó diferentes estudios acerca de su desarrollo y caracterís ticas en 1904.
* Baja estatura
* Piel seca y arrugada
* Calvicie prematura
* Canas en la infancia
* ojos prominente s
* Cráneo de gran tamaño
* Venas craneales sobresalie ntes
* Ausencia de cejas y pestañas
* Nariz grande y con forma de pico
* Mentón retraído
* Problemas cardíacos
* Pecho angosto, con costillas marcadas
* Extremidad es finas y esquelétic as
* Estrechami ento de las arterias coronarias
* Articulaci ones grandes y rígidas
* Manchas en la piel semejantes a las de la vejez por mal metabolism o de la melanina
* Presencia de enfermedad es degenerati vas como la artritis, propias de la vejez
* Muerte natural hacia los 13 años.
La progeria se produce por mutaciones en la lamina A/C las cuales generan una laminación alternativ a que lleva a la producción de una proteína inmadura similar a la prelamina A. Las células presentan un núcleo con alteracion es estructura les (herniacion es y lóbulos) así como defectos en la organizaci ón de la heterocrom atina. Molecularm ente presentan un defecto en el mecanismo de reparación del ADN como consecuenc ia de la rotura de la hélice doble.
Aunque recienteme nte se ha descubiert o específica mente el gen causante de la progeria, aún no hay cura. El promedio de vida en niños enfermos es de 13 años, pero se conoce un caso en el que el enfermo vivió hasta los 20, aunque los estudios demuestran que tenía una progeria distinta a la descrita y conocida.
Actualment e, la esperanza de vida en algunos casos se ha prolongado hasta los 21 a
No existe aún un tratamient o de probada eficacia. La mayoría de los tratamient os se limitan a paliativos o prevención de complicaci ones como son las enfermedad es cardiovasc ulares. Se utilizan aspirinas en bajas dosis y dietas hipercalór icas. Se han intentado tratamient os con hormona de crecimient o humano.
Después de ser descubiert o el gen causante de la enfermedad y su mecanismo se ha propuesto un tratamient o con un tipo de droga anticancer ígena, inhibidora de la farnesyltr ansferasa (FTIs), se ha probado su eficacia en modelos con ratones.
A partir de Mayo de 2007 se inició un período de pruebas clínicas con pacientes utilizando FTI Lonafarnib .
La verdadera Gente Pequeña (Enanismo Primordial)

El enanismo primordial es una enfermedad que al contrario del enanismo habitual, es un enanismo en todos los niveles, el cual solo lo padecen alrededor de 100 personas en el mundo.
Poco se conoce acerca de las personas más pequeñas del mundo. El enanismo primordial es el tipo más raro de enanismo que trae como consecuenc ia un tamaño de cuerpo más pequeño en todas las etapas de la vida, comenzando desde antes del nacimiento . Los niños que padecen esta enfermedad rara vez crecen más de 90 centímetro s. Se estima que existen solamente 100 individuos en el mundo con este trastorno, 40 de ellos en los Estados Unidos.
y por ultimo ,pero no menos importante (por ahora el ultimo) , han visto las cabezas reducidas ? , pues imaginense el vivir con una (y reflexione n de paso)
Microcefal ia

La microcefal ia es un trastorno neurológic o en el cual la circunfere ncia de la cabeza es más pequeña que el promedio para la edad y el sexo del niño. La microcefal ia puede ser congénita o puede ocurrir en los primeros años de vida. El trastorno puede provenir de una amplia variedad de condicione s que provocan un crecimient o anormal del cerebro o de síndromes relacionad os con anormalida des cromosómic as.
Los niños con microcefal ia nacen con una cabeza de tamaño normal o reducida. Posteriorm ente, la cabeza deja de crecer mientras que la cara continúa desarrollá ndose normalment e, lo que produce un niño con la cabeza pequeña, la cara grande, una frente en retroceso y un cuero cabelludo blando y a menudo arrugado. A medida que el niño se hace mayor, la pequeñez del cráneo llega a ser más obvia, aunque todo el cuerpo generalmen te presenta también peso insuficien te y enanismo. El desarrollo de las funciones motrices y del habla puede verse afectado. La hiperactiv idad y el retraso mental son comunes, aunque el grado de cada uno varía. También pueden ocurrir convulsion es. La capacidad motora varía, pudiendo evidenciar se desde torpeza en algunos casos hasta cuadripleg ia espástica (parálisis) en otros.
Generalmen te no existe un tratamient o específico para la microcefal ia. El tratamient o es sintomátic o y asistencia l.
En general, la esperanza de vida para los individuos con microcefal ia se reduce y el pronóstico para la función normal del cerebro es pobre. El pronóstico varía dependiend o de la presencia de ciertas anormalida des relacionad as.
Miren estos videos :S
[YOUTUBE]mwaajwHSIv8[/YOUTUBE]
[YOUTUBE]pwfdTndbNfA[/YOUTUBE]
Fuente: Colegio, Wikipedia y otros foros.
Ictiosis arlequín es una enfermedad de la piel extremadam ente rara del grupo de las llamadas genodermat osis (grupo de dermatosis hereditari as con trastornos metabólico s). Es la forma de ictiosis congénita más grave, se hace evidente ya desde el nacimiento y debe su nombre al aspecto que tienen los recién nacidos con la enfermedad, que recuerda a un disfraz de arlequín. La ictiosis tipo Arlequín es una enfermedad genética rara de la piel caracteriz ada por escamas grandes y gruesas que aparecen en toda la piel, como a su vez se nace con los párpados volteados por lo que en lugar de ojos se observan los párpados totalmente rojos. Se asocia generalmen te deformidad es faciales caracterís ticas y a menudo anomalías en otras partes del cuerpo, especialme nte en el tórax.
la enfermedad debe su nombre al aspecto que tienen los recién nacidos con la enfermedad, que recuerda a un disfraz de arlequín.
Se debe a una alteración de la queratiniz ación cuyo mecanismo fisiopatol ógico se desconoce, pero se piensa que existe una disgenesia de la capa lamelar, probableme nte debida a anomalías de los lípidos cutáneos, que da lugar a una hiperquera tosis folicular masiva.
Aparecen fisuras profundas e irregulare s que cubren la superficie corporal, ectropión (inversión hacia fuera de los párpados) y eclabium (inversión hacia fuera de los labios) debidos a la tracción mecánica que ejerce la piel engrosada sobre la conjuntiva y la mucosa oral, hipoplasia (desarrollo incompleto o defectuoso) de orejas nariz y dedos y los recién nacidos adoptan una postura semiflexio nada.
Las complicaci ones clínicas más importante s se producen a causa del fallo de la función de barrera que la piel tiene en condicione s normales e incluyen sepsis (infección o contaminac ión generaliza da) y deshidrata ción que conducen a hipernatre mia (aumento anormal de sodio en sangre) y malnutrici ón.
Los niños nacen con constricci ón marcada de tórax y abdomen con las correspond ientes dificultad es respirator ias y de alimentaci ón.
El pronóstico es malo, ya que la mayoría fallece a los pocos días o semanas del nacimiento . Aunque existen casos excepciona les en donde la persona llega a la adolescenc ia, pero necesita de tratamient os especiales ..
pasemos a lo siguiente. ...creo que muchos escuchamos la noticia hase años , de una niña peruana que habia nacido con cierta peculiarid ad , bueno , permitanme precentarl es a la....
Niña Sirena (sirenomelia o síndrome de la sirena)
La sirenomeli a (síndrome de la sirena), es una malformaci ón congénita de etiología desconocid a que se caracteriz a principalm ente por la unión de los miembros inferiores y generalmen te está asociada a otras malformaci ones digestivas, respirator ias, cardiacas, renales, etc. Las posibilida des de vida de estos niños son escasas y la mayoría fallece dentro de los primeros 7 días de vida.
La fusión de las extremidad es inferiores es conocida como Sirenomeli a y se asocia con frecuencia a alteracion es en la formación de la pelvis y sistema genitourin ario.
La única extremidad inferior contiene el normal numero de huesos fusionado y recubierto por tejido blando y piel Lo mas usual es el fémur dividido en 2 o 3 huesos y debajo de la rodilla 5 o 6 dígitos retembland o el pie.
El sacro esta ausente y el hueso iliaco esta fusionado con un simple acetábulo presente en la parte posterior de la pelvis.
El ano siempre es imperforad o, los riñones, uréteres, vejiga y uréteres están ausentes. Los órganos genitales rara vez son encontrado s.
Algunos Sirenomelu s muestran gran reducción en el desarrollo de las extremidad es inferiores, ellos no tiene piernas y malformaci ones en tronco, estos casos son incompatib les con la vida.
bueno , sigamos .
muchos de ustedes , han escuchado y visto , las hstorias de hombres lobo , bueno , en esta ocacion les traigo algo real , que , es muy ajeno a lo que ustedes pensaran (no todos , algunos)
este es......
El Hombre Lobo (Hipertricosis Congénita)
La Hipertrico sis Congénita
El Síndrome del Hombre Lobo (también llamado Hipertrico sis Universal Congénita) es un claro ejemplo de que la realidad supera con creces a la ficción.
Existen dos variantes más típicas:
Hipertrico sis Lanuginosa Congénita. Es una enfermedad extremadam ente rara ya que sólo se han documentad o 50 casos desde la Edad Media. Las personas que lo padecen están completame nte cubiertas por un vello lanugo largo excepto en las palmas de las manos y de los pies. La longitud a la cual puede llegar el vello es de 25 centímetro s.
El lanugo es el pelo fino y blanquecin o (como si fuera pelusilla) que aparece en los recién nacidos en hombros y brazos y que desaparece normalment e tras el primer mes desde el nacimiento . En los que padecen esta forma de hipertrico sis el lanugo persiste y puede crecer durante toda la vida o desaparece r con los años.
Síndrome de Ambras. En esta variante el vello es más grueso, posee coloración y en todos los casos crece a lo largo de toda la vida.
Como suele suceder con estas anomalías tan raras, apenas se han estudiado. Sólo se sabe que se deben a una mutación genética dominante. La mayoría de veces los individuos lo adquieren por herencia familiar, el hecho de que la mutación sea hereditari a hace que sea normal que existan familias con numerosos miembros que posean esta alteración . Ya que hay un 50% de probabilid ades de que el descendien te posea el síndrome. Pero otras veces aparecen mutaciones de forma espontánea . De todas formas, no se sabe la localizaci ón genética, ni cómo actúa dicha mutación.
Salvo la exagerada presencia de pelo, no sufren ninguna otra alteración . No tienen una esperanza de vida menor ni tienen mayor probabilid ad de enfermar. Sin embargo, la presencia del individuo en la sociedad hace que a menudo se vea aislado, discrimina do o maltratado física o psicológic amente, por lo que son proclives a tener serios problemas psicológic os. La sociedad es muy dura con respecto a aquello que desconoce y es diferente.
Una de las razones por las que existe la fantasía del Hombre Lobo se debe a que podría haber sido asociado a este raro síndrome. Sin embargo, se sabe que hay otras enfermedad es que también han contribuid o a difundirlo, como la porfiria, la licantropí a, el lupus eritematos o sistémico…
Asociacion es a la Leyenda del Hombre Lobo
Personajes peludos han sido con frecuencia malinterpr etados como hombres lobo, aunque en este caso más por su morfología que por sus caracterís ticas sanguinari as.
Un amplio grupo de estos los encontramo s en la historia de los llamados 'fenómenos humanos' y, por desgracia, asociados la mayoría de las veces a la exhibición circense o a un coleccioni smo arcaico y de dudoso gusto por parte de nobles y monarquías por suerte obsoletas. El más antiguo conocido hace referencia a un canario nacido en 1556 llamado Petrus Gonsalvus, que tenía todo el cuerpo y la cara cubiertos de vello. Por orden del rey Enrique II de Francia, se transladó a París donde tuvo una exquisita educación y tuvo 4 hijos, todos con el mismo aspecto de su padre. Tras una gira por Europa de toda la familia, el duque Albretch IV de Babiera asombrado por su aspecto encargó realizarle un retrato de tamaño natural, obra que regaló posteriorm ente al archiduque Ferdinand del Tirol y que expuso en su Castillo de Ambras, en Innsbruck, Austria. Posteriorm ente la artista Lavinia Fontana de Zappis realizó otro famoso cuadro de su hija Antonietta .
Al ser el primer caso detallado del que se tuvo noticia, este tipo de hipertrico sis universal congénita es conocida como el Síndrome de Ambras -una de las llamadas enfermedad es raras con inversión en el cromosoma 8 (p11.2q23.1)-, que curiosamen te nos ofrece una imagen muy similar al clásico hombre lobo cinematogr áfico.
Retrato de Petrus Gonsalvus, nacido en 1556 quien se hizo famoso como espectácul o entre la corte de la época por tener todo el cuerpo y cara cubiertos de vello.
Barbara Urselin fue otro famoso caso de grave hipertrico sis del que también existe testimonio gráfico. Nació en 1629 en Kempten, Alemania, y fue exhibida de muy pequeña por sus padres a cambio de dinero como "La Mujer Cubierta de Pelo" (The Hairy-Faced Woman); posteriorm ente se casó y su marido continuó con este dudoso negocio recorriend o toda Europa. También se tiene constancia pictórica del siglo XVII, obra de Stefano della Bella, de un tal Horacio González, un hirsuto hombre lobo español que viajó a Roma en peregrinac ión para rogar el milagro de un cambio de aspecto.
Otros famosos personajes peludos fueron Adrian Jefticheje v, conocido en 1873 como "El Hombre Salvaje de los Bosques de Kostroma" y que se exhibía como el fruto de las relaciones entre un oso y una campesina, de carácter arisco y de pésimo humor agravado por una hepatopatí a debida a su aficción al vodka, así como su hijo ilegítimo Fedor, conocido posteriorm ente por el nombre de Theodore Petroff y por el sobrenombr e de "Jo-Jo, el Niño con Cara de Perro", "El Skye Terrier Humano" o "El Hombre Caniche", y que gracias a su ingenio y buen carácter fue muy querido por el público y la prensa que lo visitaba en el circo. Tras su muy llorada muerte en 1904, el circo buscó un fenómeno similar encontrand o a Stephan Bibrowski, conocido como "Lionel, el Hombre León", que había nacido en 1891 de padres normales; personaje culto y divertido, falleció en 1931 tras una exitosa carrera como showman.
Entre 1888 y 1889, una familia de birmanos que huyeron de su país durante la guerra civil, hizo una gira por Estados Unidos; el padre llamado Shwe-Maong, peludo como los casos anteriores, fue regalado de niño al rey como curiosidad y divertimen to de palacio; tras casarse, tuvo cuatro hijas, una de ella peluda y que posteriorm ente tuvo un hijo normal y una hija también peluda. Y si revisamos la literatura médica, nos encontrare mos ocasionalm ente con este síndrome: un hombre chino en 1937, un alemán en 1958, otra alemana en 1964, un niño griego en 1993 y toda una familia mexicana con 21 miembros afectados y que son conocidos como "Los Niños Lobo".
La medicina también ha encontrado otro tipo de hirsutismo congénito diferente al que hemos visto; en este caso también puede afectar a todo el cuerpo pero se asocia a un prognatism o facial, hipertrofi a gingival y alteracion es dentales. Este síndrome de hirsutismo con fibromatos is gingival se ha asociado más a una imagen de oso o de mono, que a un perro o a un lobo, pero no podemos evitar introducir lo aquí por su similitud.
Ejemplos famosos de este síndrome ha sido el trágico caso de Julia Pastrana, una india mexicana que nació en 1834 en las montañas de Sierra Madre; a los 20 años se puso a trabajar como fenómeno profesiona l exhibiéndo se por la región y los EEUU. Era una mujer baja, de 137 cm de altura, con una gran hipertrofi a gingival que formaba unas grandes encías llenas de protuberan cias, con la frente muy peluda y unos bigotes y barba muy llamativos . Se le presentó como "El Híbrido Maravillos o" o "La Mujer Oso", y muchos creyeron que era el resultado de los bestiales amoríos de un humano y una osa o un orangután. Su vida, por desgracia, estuvo rodeada por los poquísimos escrúpulos de su mánager, que se casó con ella para poder seguir explotando el filón de la curiosidad humana; la dejó embarazada y llegó a vender entradas para asistir al parto. Julia dió a luz en 1860 a un niño tan peludo como ella pero, debido a lo dificultos o del alumbramie nto, falleció a los tres días y la madre dos días después. El marido continuó aprovechán dose de ambos y los mandó embalsamar (aunque en realidad fue un puro trabajo de taxidermia) y siguió exhibiéndo los por el mundo. Tras muchas peripecias las exhibicion es terminaron en 1976 cuando unos ladrones destruyero n parcialmen te sus momias; desde el año 1990 sus restos se encuentran en el Instituto de Medicina Forense Rikshospit alet de Oslo, donde sólo están disponible s para estudios científico s.
Barbara Urselin quien se hizo famosa como atracción circense y una familia birmana de "hombres lobo", como se puede observas los hijos heredaron la anomalía.
Otra caso que podríamos incluir en este grupo sería el de Krao, una niña hirsuta con un pelo corporal negro, lacio y lustroso, con prognatism o facial, nariz chata y orejas grandes, ágil de movimiento s y muy flexible. Al igual que otras personas de caracterís ticas similares fue exhibida desde 1883 por Europa y EEUU como "La Mujer Simio" junto con una falsa historia familiar que la trasformab a en una especie de eslabón perdido, aunque nació en realidad en Bangkok de padres normales.
También podríamos incluir aquí el famoso caso español de la llamada "Osa de Andara", una mujer velluda de Cantabria estudiada en 1875, descrita con pelo crespo, de frente aplastada y estrecha, nariz chata, pómulos prominente s y labios parecidos a un hocico. Algún autor afirmó, con pocos visos de realidad, que era una pastora llamada Joaquina López que huyó a las cuevas de Andara avergonzad a por su imagen; otros, por el contrario, han intentado ver en esta mujer un resto de la raza neandertal ...
Hombre de Piedra (Fibrodisplasia osificante progresiva)
Se llama Carlos Olea tiene 44 años, dice sentir "algo por dentro, como si los nervios se estuvieran poniendo duros" y es conocido en Chile como "el hombre de piedra".
Este chileno padece desde su nacimiento fibrodispl asia osificante progresiva, una rara patología genética que ha destruido sus músculos y sus articulaci ones, por lo que su movilidad es casi nula.
nfermedad de Munchmeyer, esqueletog énesis ectópica o fibrodispl asia osificante progresiva (FOP) son algunos de los nombres dados a esta patología que provoca una miositis osificante de ciertos tejidos blandos, es decir, partes del cuerpo como músculos o tendones se osifican, se convierten en hueso.fect a, según algunos estudios, a unas 2500 personas aproximada mente en todo el mundo.
El 75% de la población afectada suele presentar al nacer deformidad es que pueden ayudar al reconocimi ento de la enfermedad . Las malformaci ones más comunes son la microdacti lia y la anquilosis en las falanges de los dedos gordos del pie.
Estas formacione s óseas se producen por brotes desde la infancia. Tras la inflamació n de los tejidos blandos en cuestión (músculos tendones o ligamentos), éstos se convierten en hueso, lo que poco a poco supone la pérdida de movilidad. Los primeros signos de la enfermedad se suelen dar en las zonas cercanas a la columna dorsal y posteriorm ente se van propagando hacia otras articulaci ones y grupos musculares como los codos o las rodillas. El avance de la patología es progresivo, según se desarrolla aparecen deformidad es, discapacid ades funcionale s y perturbaci ones conductual es. Por otro lado tanto los músculos faciales, como musculos indispensa bles para funciones vitales como lo es para la respiració n el diafragma o el corazón para la circulació n sanguínea, están caracterís ticamente fuera de peligro.
Desde el siglo XIX ha habido referencia s esporádica s en la literatura médica describien do a pacientes que, aparenteme nte, "se volvieron piedra". Es posible que algunos de estos casos pueda haber sido atribuible a la FOP.
Quizá el caso más conocido de FOP en tiempos modernos sea el de Harry Raymon Eastlack Jr., nacido en Filadelfia, PA en noviembre de 1933. Su condición comenzó a desarrolla rse a la edad de diez años y para la época de su muerte por una neumonía en noviembre de 1973 (seis días antes de su cuadragési mo cumpleaños), su cuerpo se había osificado por completo y sólo podía mover sus labios.
Lo que hace al caso de Eastlack particular mente notable es que antes de su muerte hizo conocer su intención de donar su cuerpo a la ciencia, con la esperanza de que su muerte pueda ayudar a encontrar una cura a esta poco entendida y particular mente cruel enfermedad . Como era su deseo, su esqueleto preservado ahora reside en el museo Mütte
en el Colegio de Medicina de Filadelfia y fue una invalorabl e fuente de informació n para el estudio de esta enfermedad .
Munchmeyer describe la enfermedad en 1969, aunque la primera mención del cuadro patológico fue hecha por Guy Patin en 1992.
Según estudios de la Universida d de Pensilvani a la causa se halla en la activación de un gen (el ACVR1) mutado, que provoca la formación de tejido cartilagin oso u óseo en las zonas en las que se encuentra. No hay tratamient o ni medicación que sea 100% eficaz contra esta condición.
esto ya se los habia explicado antes , pero espero que recuerden bien a...
Los Bebes Ciclope (holoprosencefalia y cliclopia)
La holoprosen cefalia constituye un amplio espectro de malformaci ones del cráneo y la cara debidas a una anormalida d compleja del desarrollo del cerebro que tienen en común la ausencia del desarrollo del prosencéfa lo, que es el lóbulo frontal del cerebro del embrión.[2] Durante el desarrollo normal se forma el lóbulo frontal y la cara comienza a desarrolla rse en la quinta y sexta semana del embarazo. La holoprosen cefalia es causada por la falta de división del lóbulo frontal del cerebro del embrión para formar los hemisferio s cerebrales bilaterale s (las mitades izquierda y derecha del cerebro), causando defectos en el desarrollo de la cara y en la estructura y el funcionami ento del cerebro... eso es la Holopresen cefalia
La ciclopía es una malformaci ón congénita poco frecuente, producto de una secuencia que se inicia con un defecto en la división del cerebro embrionari o que conlleva a holoprosen cefalia alobar y la fusión de los ojos en un único ojo central.
Este es un caso de un recien nacido con ciclopía producto de embarazo de madre de 19 años, G2 A1, quien ingresa remitida por malformaci ones congénitas múltiples, en trabajo de parto, con embarazo de 30 semanas.
Se practicó cuatro ecografías, la primera a las 20 semanas en la que no se observaron malformaci ones; la segunda a las 28 semanas, en la que se encuentra microcefal ia y un quiste en el hemisferio derecho, por lo cual se solicita ecografía de detalle anatómico, encontrand o holoprosen cefalia alobar, ventrículo único, anoftalmía, micrognati a, ausencia de puente nasal, clinodacti lia y polihidram nios. Asistió a tres controles prenatales .
En el primer trimestre se embriagó en dos oportunida des y se aplicó tres ampollas de diclofenac o. Por vía vaginal nace RN muerto.
En la autopsia se encontró un único lóbulo cerebral, con un ventrículo, dos órbitas parcialmen te fusionadas, un globo ocular con un cristalino y un nervio óptico. No se halló nariz. También se encontraro n dos conductos lacrimales . Cariotipo 46, XX.
y este es el ultimo (por ahora) pero no menos importante .....
El Hombre Elefante (sindrome de Proteus)
El Síndrome de Proteus es una enfermedad congénita que causa un crecimient o excesivo de la piel y un desarrollo anormal de los huesos, normalment e acompañado s de tumores en más de la mitad del cuerpo.
El Síndrome de Proteus es extremadam ente raro. Desde que el Dr. Michael Cohen lo identificó en 1979, sólo se han confirmado poco más de 200 casos en todo el mundo. Puede que se hayan dado más, pero los que son correctame nte diagnostic ados suelen tener manifestac iones evidentes del síndrome, estando considerab lemente desfigurad os.
Esta extraña enfermedad habría permanecid o oculta de no ser por que Joseph Merrick – inmortaliz ado como el "Hombre Elefante" debido al aspecto que le daban los tumores de su cabeza y el color grisaceo de su piel – fue diagnostic ado más tarde de un severo caso de Síndrome de Proteus además de, neurofibro matosis, cosa que los médicos pensaban anteriorme nte que tenía. Extrañamen te, el brazo izquierdo y los genitales de Merrick no estaban afectados por la enfermedad que había deformado el resto de su cuerpo.
El Síndrome de Proteus causa un crecimient o anormal de la piel, huesos, músculos, tejido adiposo, y vasos sanguíneos y linfáticos .
Es una enfermedad progresiva, los niños suelen nacer sin ninguna deformidad evidente. Conforme crecen aparecen los tumores y el crecimient o de la piel y de los huesos. La gravedad y la localizaci ón de estos crecimient os asimétrico s varían ampliament e, aunque suelen darse en el craneo, uno o más miembros y en la plantas de los pies. Hay un riesgo de muerte prematura en los individuos afectados debido a trombosis y tromboembo lismo pulmonar, causadas por malformaci ones asociadas a este desorden en los vasos. Otros riesgos pueden venir provocados por el peso del tejido extra- Se cree que Joseph Merrick murió por el peso de su enorme y pesada cabeza que venció la resistenci a de su cuello y cayó hacia atrás, fracturánd oselo.
El desorden afecta a los dos sexos por igual, y puede darse en todas las etnias.
Esqueleto y craneo de Merrick
bueno ,por ahora esto a sido todo tipos , espero haya sido de su agrado , este es para mi uno de mis mejores temas hasta ahora (por favor no sean ogts tipos )...
Niños Ancianos (Progeria o Síndrome de Werner)
Progeria (del griego geras, "vejez") es una enfermedad genética de la infancia extremadam ente rara, caracteriz ada por un envejecimi ento brusco y prematuro. Se estima que afecta a uno de cada 8 millones de recién nacidos.
La forma más severa de esta enfermedad es la llamada síndrome de Hutchinson-Gilford nombrada así en honor de Jonathan Hutchinson, quién fue el primero en describirl a en 1886 y de Hastings Gilford quien realizó diferentes estudios acerca de su desarrollo y caracterís ticas en 1904.
sus caracteris ticas clinicas son:
Progeria (del griego geras, "vejez") es una enfermedad genética de la infancia extremadam ente rara, caracteriz ada por un envejecimi ento brusco y prematuro. Se estima que afecta a uno de cada 8 millones de recién nacidos.
La forma más severa de esta enfermedad es la llamada síndrome de Hutchinson-Gilford nombrada así en honor de Jonathan Hutchinson, quién fue el primero en describirl a en 1886 y de Hastings Gilford quien realizó diferentes estudios acerca de su desarrollo y caracterís ticas en 1904.
* Baja estatura
* Piel seca y arrugada
* Calvicie prematura
* Canas en la infancia
* ojos prominente s
* Cráneo de gran tamaño
* Venas craneales sobresalie ntes
* Ausencia de cejas y pestañas
* Nariz grande y con forma de pico
* Mentón retraído
* Problemas cardíacos
* Pecho angosto, con costillas marcadas
* Extremidad es finas y esquelétic as
* Estrechami ento de las arterias coronarias
* Articulaci ones grandes y rígidas
* Manchas en la piel semejantes a las de la vejez por mal metabolism o de la melanina
* Presencia de enfermedad es degenerati vas como la artritis, propias de la vejez
* Muerte natural hacia los 13 años.
La progeria se produce por mutaciones en la lamina A/C las cuales generan una laminación alternativ a que lleva a la producción de una proteína inmadura similar a la prelamina A. Las células presentan un núcleo con alteracion es estructura les (herniacion es y lóbulos) así como defectos en la organizaci ón de la heterocrom atina. Molecularm ente presentan un defecto en el mecanismo de reparación del ADN como consecuenc ia de la rotura de la hélice doble.
Aunque recienteme nte se ha descubiert o específica mente el gen causante de la progeria, aún no hay cura. El promedio de vida en niños enfermos es de 13 años, pero se conoce un caso en el que el enfermo vivió hasta los 20, aunque los estudios demuestran que tenía una progeria distinta a la descrita y conocida.
Actualment e, la esperanza de vida en algunos casos se ha prolongado hasta los 21 a
No existe aún un tratamient o de probada eficacia. La mayoría de los tratamient os se limitan a paliativos o prevención de complicaci ones como son las enfermedad es cardiovasc ulares. Se utilizan aspirinas en bajas dosis y dietas hipercalór icas. Se han intentado tratamient os con hormona de crecimient o humano.
Después de ser descubiert o el gen causante de la enfermedad y su mecanismo se ha propuesto un tratamient o con un tipo de droga anticancer ígena, inhibidora de la farnesyltr ansferasa (FTIs), se ha probado su eficacia en modelos con ratones.
A partir de Mayo de 2007 se inició un período de pruebas clínicas con pacientes utilizando FTI Lonafarnib .
La verdadera Gente Pequeña (Enanismo Primordial)
El enanismo primordial es una enfermedad que al contrario del enanismo habitual, es un enanismo en todos los niveles, el cual solo lo padecen alrededor de 100 personas en el mundo.
Poco se conoce acerca de las personas más pequeñas del mundo. El enanismo primordial es el tipo más raro de enanismo que trae como consecuenc ia un tamaño de cuerpo más pequeño en todas las etapas de la vida, comenzando desde antes del nacimiento . Los niños que padecen esta enfermedad rara vez crecen más de 90 centímetro s. Se estima que existen solamente 100 individuos en el mundo con este trastorno, 40 de ellos en los Estados Unidos.
y por ultimo ,pero no menos importante (por ahora el ultimo) , han visto las cabezas reducidas ? , pues imaginense el vivir con una (y reflexione n de paso)
Microcefal ia
La microcefal ia es un trastorno neurológic o en el cual la circunfere ncia de la cabeza es más pequeña que el promedio para la edad y el sexo del niño. La microcefal ia puede ser congénita o puede ocurrir en los primeros años de vida. El trastorno puede provenir de una amplia variedad de condicione s que provocan un crecimient o anormal del cerebro o de síndromes relacionad os con anormalida des cromosómic as.
Los niños con microcefal ia nacen con una cabeza de tamaño normal o reducida. Posteriorm ente, la cabeza deja de crecer mientras que la cara continúa desarrollá ndose normalment e, lo que produce un niño con la cabeza pequeña, la cara grande, una frente en retroceso y un cuero cabelludo blando y a menudo arrugado. A medida que el niño se hace mayor, la pequeñez del cráneo llega a ser más obvia, aunque todo el cuerpo generalmen te presenta también peso insuficien te y enanismo. El desarrollo de las funciones motrices y del habla puede verse afectado. La hiperactiv idad y el retraso mental son comunes, aunque el grado de cada uno varía. También pueden ocurrir convulsion es. La capacidad motora varía, pudiendo evidenciar se desde torpeza en algunos casos hasta cuadripleg ia espástica (parálisis) en otros.
Generalmen te no existe un tratamient o específico para la microcefal ia. El tratamient o es sintomátic o y asistencia l.
En general, la esperanza de vida para los individuos con microcefal ia se reduce y el pronóstico para la función normal del cerebro es pobre. El pronóstico varía dependiend o de la presencia de ciertas anormalida des relacionad as.
Miren estos videos :S
[YOUTUBE]mwaajwHSIv8[/YOUTUBE]
[YOUTUBE]pwfdTndbNfA[/YOUTUBE]
Fuente: Colegio, Wikipedia y otros foros.

